Leur exposition face aux facteurs externes
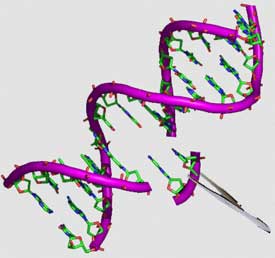
Les plantes génétiquement modifiées ont ici été conçues pour résister à des attaques extérieures, attaques qui nuisent aux rendements et parfois même à l’environnement. Nous allons ici voir les trois attaques extérieures principales que subissent les plantes c’est-à-dire : les herbicides, les insectes ainsi que les virus.
Les herbicides sont très polluants, les insectes se nourrissent des plantes et les virus contaminent les plantes entraînant une baisse de rendements.
Les herbicides
Les herbicides (aussi appelés désherbants) sont des produits chimiques appartenant à la famille des pesticides. Ils sont utilisés par les fermiers afin de se débarrasser des mauvaises herbes. Par exemple, une plante, une fois génétiquement modifiée, pourra être résistante au glyphosate. Le glyphosate est l’ingrédient actif de plusieurs herbicides vendus partout dans le monde dont le fameux Roundup.
Il existe différents types de tolérance à un herbicide. Les plantes génétiquement modifiées utilisent habituellement l’une des deux méthodes suivantes :
- La plante transgénique produit une nouvelle protéine* annulant l’effet toxique de l’herbicide.
- La protéine normalement ciblée par l’herbicide est remplacée dans la plante génétiquement modifiée par une nouvelle protéine non sensible à l’herbicide.
Par ailleurs, aux Etats-Unis, plus de 90% du maïs, soja et coton sont résistants à un herbicide.
Si la plante est immunisée à un herbicide précis, alors l’agriculteur peut épandre* l’herbicide dans le champs. Les plantes transgéniques vont survivre contrairement aux mauvaises herbes.
Les insectes
Les insectes dévastaient les cultures auparavant. C’est pour cela que les agriculteurs utilisent des insecticides qui tuent les espèces ciblées mais qui sont polluants. Néanmoins, désormais il existe une autre alternative étant la modification génétique. Elle permet à la plante de sécréter son propre insecticide. Ces plantes transgéniques sont appelées des plantes «Bt» ce qui provient de Bacillus Thuringiensis (bactérie se trouvant dans la nature). Elle a été implantée dans le génome des plantes. Il existe plusieurs variantes de toxines naturelles produites par la bactérie Bt qui vont permettre de lutter contre différentes espèces d’insectes en les ciblant. Par exemple, la toxine Cry1Ab, l’une des plus utilisées, permet de tuer les lépidoptères (papillons de jour et nuit).
Prenons le cas du maïs Bt qui est une variété de maïs génétiquement modifiée. Utiliser ce type de maïs permet tout d’abord l’abondance des récoltes mais aussi la diminution de l’usage des produits chimiques. Ainsi, ce maïs génétiquement modifié constitue un autre moyen pour les agriculteurs que les insecticides pour lutter contre les insectes nuisibles. Cette variété existe depuis 2003 et constitue 65% des cultures de maïs aux Etats-Unis. Un gène a été ajouté au maïs afin de résister aux insectes nuisibles comme c’est le cas des pyrales (papillons dont les chenilles l’infestent parfois de manière massive, voir photographie ci-contre). La plante sécrète alors une protéine toxique (provenant de la bactérie Bt ayant des propriétés insecticides) afin d’éliminer la pyrale. La protéine Cry1Ab a été introduite dans le génome* de certaines variétés de mais afin de les rendre résistantes à l’insecte ravageur. Le gène permet l’expression de cette protéine.
Illustration 4 : Photographie de la pyrale du maïs :

Dans des états d’Amérique tels que le Wisconsin et l’Iowa, l’utilisation du maïs Bt (entre 1996 et 2009) a permis de réduire la population de pyrales jusqu’à 73%.
D’ailleurs, la bactérie Bacillus thuringiensis est considérée sans danger pour l’être humain étant donné que notre organisme digestif la digère rapidement empêchant de faire effet.
En plus de se nourrir des cultures, les insectes transmettent parfois des virus aux plantes ce qui nous mène à notre dernière résistance : celle face aux virus.
Les virus
Plusieurs plantes sont sujettes à des maladies virales. Les virus sont des agents infectieux très petits possédant un seul type d’acide nucléique : l’ADN ou l’ARN qui ne peut se reproduire qu’en parasitant une cellule. Cet acide nucléique est entouré d’une enveloppe de protéines.
Illustration 5 : Schéma de la structure d’un virus :
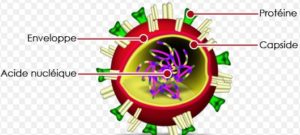
La capside est la couche de protéines recouvrant l’acide nucléique du virus.
Les virus constituent les attaques les plus nocives pour les plantes étant donné aucun traitement curatif* n’existe. Ils sont donc à l’origine de nombreux dégâts sur les cultures entraînant une importante perte de rendements. Tout d’abord, il existe une méthode mécanique permettant d’enlever les plantes atteintes du virus. Cependant, pour résoudre ce problème, nous allons nous intéresser aux plantes résistantes aux virus après modification génétique. Lorsqu’un virus infecte une cellule il y a une synthèse* et plusieurs copies de son enveloppe de protéines. Le virus se reproduit alors ce qui infectera les cellules avoisinantes.
Les plantes résistantes aux virus réalisent une synthèse des protéines bloquant la multiplication des virus ainsi que leur développement.
Ainsi, les plantes, après avoir subi une modification génétique, peuvent être résistantes à diverses attaques extérieures (herbicides, insectes et virus). Nous allons désormais aborder cette modification génétique, plus connue sous le nom de transgenèse.
Le principe de la transgenèse
La première manipulation génétique, comme nous l’avons vu dans la première partie, a lieu en 1972 grâce à Paul Berg et son équipe.
La transgenèse est l’ensemble des techniques permettant de transférer un ou plusieurs gènes d’un organisme «donneur» à un organisme «receveur». Elle est notamment utilisée en médecine et dans l’industrie. Cependant, ce qui nous intéresse ici est son usage dans l’agriculture. Elle permet à l’organisme receveur de produire une nouvelle protéine grâce au gène qui lui a été introduit. La possibilité d’introduire un gène à tout organisme s’explique par le fait que l’ADN soit universel. En effet, les gènes sont codés, lus et traduits de la même façon chez tous les organismes. L’organisme receveur étant génétiquement modifié, il s’agit donc d’un OGM.
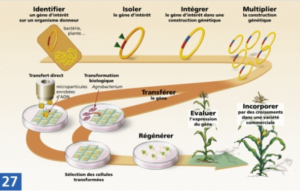
Un plant de maïs se fait attaquer quotidiennement par des insectes, un apprenti agriculteur souhaite y remédier mais ne sait pas comment procéder. Nous nous dévouons alors et lui proposons de réaliser les étapes suivantes :
- Tout d’abord, nous devons identifier le gène d’intérêt c’est-à-dire le gène dont les caractéristiques sont intéressantes pour l’organisme que nous souhaitons modifier (le plant de maïs) dans notre situation (plant attaqué par des ravageurs). Nous décidons alors d’extraire le gène d’une bactérie produisant des protéines insecticides. Nous voulons donc transférer ce gène dans le plant de maïs qui pourra alors résister aux insectes.
- Ensuite, après avoir isolé le gène d’intérêt de notre organisme donneur (la bactérie), nous l’extrayons grâce à des enzymes de restriction* qui permettent de couper l’ADN en des points précis.
Cependant, nous devons multiplier ce gène puisqu’un ne suffit pas : il en faut plusieurs milliards. - C’est alors que nous démultiplions le gène d’intérêt grâce à la technique de PCR (Polymerase Chain Reaction) ce qui signifie, en français, Réaction en Chaîne par Polymérase. Le but étant de dupliquer* notre séquence d’ADN.
Nous expliquons désormais à l’apprenti agriculteur comment transférer le gène d’intérêt dans notre plant de maïs à travers les deux méthodes de transgenèse les plus courantes.
La transgenèse par Agrobacterium Tumefaciens
- Action naturelle de la bactérie :
La bactérie est attirée par des composés phénoliques dégagés par les plantes (dicotylédones) lorsqu’elles sont blessées. L’Agrobacterium, bactérie présente dans les sols, va alors se fixer sur les cellules de la plante (grâce à la blessure formée). Cela va créer une multiplication des cellules de la plante à l’origine d’une tumeur généralement située au collet, d’où le nom de la maladie : la galle du collet. Ces cellules créées vont alors libérer des composés chimiques particuliers, les opines qui vont servir de de source d’énergie pour les bactéries à proximité.
Illustration 6 : Schéma du développement de la galle du collet :
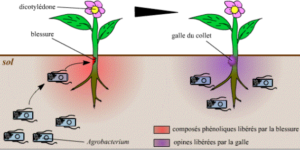
Depuis 1974, les chercheurs ont compris que ce phénomène est dû à un transfert d’une partie de l’ADN plasmidique (ADN circulaire de petite taille) de la bactérie jusque dans le génome des cellules de la plantes. Cependant, c’est plusieurs années plus tôt, en 1907 que la bactérie est identifiée à partir de tumeurs par E.F Smith et C.O Townsend, deux chercheurs américains.
Une fois ce mécanisme connu, il a été détourné dans un but de transgenèse. Pour cela, il faut remplacer l’ADN-T par un autre ADN portant notre gène d’intérêt. Il faut donc préalablement neutraliser la maladie que la bactérie peut inoculer*.
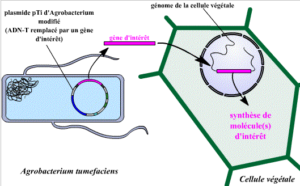
Explications des couleurs sur le plasmide pTi d’Agrobactérium :
- En rose : l’ADN-T (gène d’intérêt).
- En bleu foncé : la région virulente. Cette région comporte une série de gènes, qui permettent la fixation de la bactérie aux cellules végétales et le transfert de l’ADN-T.
- En vert : la région de catabolisme des opines. Cette région permet à la bactérie d’utiliser les opines libérées par le végétale suite à son infection par l’ADN-T.
- En bleu ciel : la région de réplication. Cette région permet au plasmide de se multiplier dans la bactérie.
Pour réaliser la transgenèse, les chercheurs vont remplacer l’ADN-T par un ADN comportant en particulier le gène d’intérêt (GI) et un gène de sélection (GS). Ce dernier va permettre de récupérer plus facilement les cellules qui ont intégrées l’ADN transgénique à leur génome. Ce gène d’intérêt, une fois présent dans le génome de la cellule végétale, va synthétiser une ou plusieurs molécule(s) d’intérêt.
La transgenèse par biolistique :
Connaissances préalables permettant de comprendre la technique biolistique :
Illustration 8 : Schéma d’une cellule végétale :
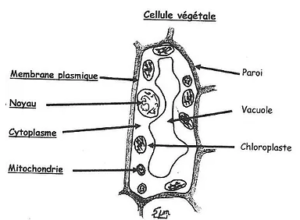
La cellule est la plus petite unité du vivant. En effet, elle est de l’ordre de plusieurs micromètres (sur le schéma il s’agit de 5 µm).
Les végétaux sont eucaryotes. Comme nous le percevons, ils possèdent un noyau dans leurs cellules contrairement aux bactéries qui sont des organismes procaryotes. C’est dans le noyau que se trouvent les chromosomes (et donc l’ADN), porteurs des caractères héréditaires. Le cytoplasme est la partie interne de la cellule constituée majoritairement d’eau et de protéines. La membrane plasmique est une enveloppe entourant la cellule et séparant le cytoplasme du milieu extérieur.
Les chloroplastes sont les organites* cellulaires des plantes de couleur verte dans lesquelles se trouve la chlorophylle. Tous les végétaux ne contiennent pas de chloroplastes dans leurs cellules: il s’agit seulement des végétaux chlorophylliens tel que le phytoplancton par exemple.
La vacuole est une enclave inerte se formant dans le cytoplasme de la cellule. La paroi est une structure formant un cadre autour de la membrane plasmique. Les mitochondries sont des organites cellulaires dans lesquelles se déroulent la respiration.
Illustration 9 : Schéma de la méthode biolistique :
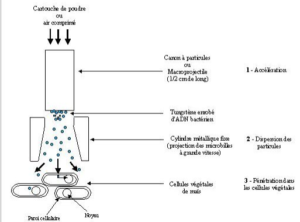
Tout d’abord, il faut enrober d’ADN des micro-billes d’or, de platine ou de tungstène d’un micron de diamètre. Notre gène d’intérêt (se trouvant dans l’ADN) est propulsé dans les cellules de la plante (maïs) à grande vitesse grâce à un canon à particules ou macroprojectile. Nous espérons désormais que parmi nos milliers de micro-billes, l’une d’entre elles transperce la cellule végétale en traversant la membrane plasmique ainsi que le cytoplasme pour arriver au noyau et se greffer à l’endroit souhaité dans l’ADN. Dans ce cas, l’ADN ainsi transféré est libéré et peut alors s’intégrer au génome* de la cellule hôte.
Après avoir effectué l’une des deux transgenèse, il faut cultiver les cellules dans un milieu sélectif* pour supprimer celles qui n’ont pas reçu le transgène* et ne garder que celles qui ont bien intégré le gène d’intérêt.
Illustration 10 : Récapitulatif des étapes de la création d’une plante transgénique :
Cependant, il existe d’autres techniques de transgenèse : la transgenèse par micro-injection, la transduction et l’électro-poration.
Actuellement, il existe encore d’autres méthodes que la transgenèse pour parvenir à modifier des organismes ce qui change donc la manière de créer des OGM. En effet, on ne cherche cette fois pas à transférer un gène d’un organisme à un autre mais à provoquer des mutations* ciblées dans le génome de la plante. Ces technologies se regroupent sous le nom de : New Plant Breeding Techniques (Nouvelles Techniques de Sélection des Plantes).
Ainsi, nous avons vu comment modifier génétiquement un organisme à travers deux méthodes de transgenèse : la transgenèse par Agrobacterium Tumefaciens et la biolistique. Désormais, il est intéressant de savoir si un organisme est génétiquement modifié par la détection d’OGM.
Détection d’OGM
Dans le cadre de nos Travaux Personnels Encadrés (TPE), nous sommes allées un jour et demi (du 15/01/18 au 16/01/18) dans un laboratoire en étant supervisées par une ingénieure.
L’objectif de cette sortie pédagogique était de réaliser une expérience permettant de détecter la modification du génome d’un organisme vivant. L’équipe que nous avons été amenée à rencontrer au laboratoire est spécialisée dans les cellules souches et biothérapies. Elle est composée de douze personnes et constituée d’un directeur, de chercheurs et enseignants chercheurs, d’ingénieurs et techniciens et de doctorants ainsi que de post-doctorants. Les recherches qu’ils effectuent se concentrent sur la thématique de la santé publique (étant donné notamment l’accroissement des pathologies cardiovasculaires à l’origine de nombreux décès). Pour cela, ils s’appuient sur des organismes qu’ils modifient génétiquement afin d’étudier les maladies et d’y trouver des remèdes. Nous allons donc présenter les expériences réalisées qui seront accompagnées de photographies prises dans le laboratoire.
L’extraction d’ADN (Acide Désoxyribonucléique)
Tout d’abord, nous disposons d’un échantillon de tissus chacune. En effet, nous souhaitons extraire l’ADN de cet élément. Le but étant de savoir, par la suite, s’il y a eu mutation ou non dans le génome de l’organisme.
JOUR 1 :
Nous déposons dans un tube un morceau de 6 à 8 mm de tissus. Nous ajoutons, à l’aide d’une pipette de précision (avec un embout stérile afin de ne pas contaminer notre échantillon), 700 μl de solution tampon (constituée de Tris 50mM pH 8, EDTA 100mM, chlorure de sodium (NaCl 100mM) et de SDS 1% qui sont des détergents). Le tampon va donc permettre à la solution de garder son pH afin que l’ADN ait le même pH que lorsqu’il se trouve dans l’organisme dont il est extrait. Nous ajoutons ensuite 12 μl de protéinase K solution (20 mg/ml). Nous plaçons nos tubes à une température de 55°C pendant toute la nuit.
JOUR 2 :
En reprenant nos tubes, nous constatons que notre morceau de tissus a disparu. En effet, il a été dissous puisqu’il ne reste que les poils et le cartilage. Nous passons le tube sur le vortex (cf. illustration 1) durant 10 secondes puis à la mini-centrifugeuse (cf. illustration 2) qui a permis de séparer la solution avec l’ADN des poils restants. Ces derniers sont donc collés au fond du tube (culot) en raison de son importante vitesse de rotation. Nous ajoutons 150 μl de chlorure de sodium (NaCl 6M saturé). Puis, nous passons la solution au vortex et à la centrifugeuse (cf. illustration 3) 15’ à RT à la vitesse maximale. Nous transférons alors le SN dans un tube propre. Le «SN» ou «surnageant» est le liquide se trouvant au-dessus du culot lorsque l’on centrifuge.
Nous ajoutons désormais 700 μl d’isopropanol dans les tubes contenant notre ADN (présent dans le tissus). L’isopropanol est un alcool permettant la précipitation de l’ADN qui se pelotonne en sa présence. Nous le passons au vortex jusqu’à ce que nous apercevons un filament d’ADN.
Puis, nous le passons à la centrifugeuse 10’ à RT à vitesse maximale. La centrifugeuse, en raison de sa vitesse de rotation très élevée a permis l’apparition d’un culot au fond du tube (le culot étant cette fois l’ADN). Nous versons alors le tube pour ne garder que le SN et laissons sécher durant quelques minutes notre tube sur du papier absorbant. Nous ajoutons 150 μl d’un alcool, l’éthanol (EtOH 70%) afin de bien nettoyer la solution. Ensuite, nous le passons au vortex et à la centrifugeuse (10 minutes). Nous retirons alors l’éthanol par renversement et laissons le tube ouvert à 55°C durant 10 minutes. Il est important de l’enlever entièrement sinon cela va poser problème pour la PCR (notre prochaine expérience menant vers la détection d’une modification génétique). Nous ajoutons 300 μl d’eau free (eau sans aucune impureté donc purifiée) et passons 10 minutes à 55°C puis au vortex et enfin nous le plaçons à 37°C durant 2 heures.
Enfin, nous conservons notre tube à 4°C jusqu’à la PCR, expérience que nous avons également réalisée et que nous allons expliquer tout de suite.
La PCR («Polymerase Chain Reaction» ou Réaction en Chaîne par Polymérase
La PCR est une technique scientifique de réplication de l’ADN dite «in vitro». Elle a été inventée en 1983 par Kary Mullis, une chercheuse californienne. Elle permet l’obtention, à partir d’un échantillon peu abondant, d’importantes quantités d’ADN. L’expérience consiste en une succession de réplications d’une matrice double brin d’ADN. Pour réaliser ces multiplications, des étapes de transitions de température sont effectuées. Chaque cycle est composé de trois étapes.
Tout d’abord, le fragment d’ADN est mis à température ambiante afin qu’il puisse mener à bien la transition de son état comprimé à sa formation de double hélice.
Photographies de l’expérience :
Illustration 11 : Préparation du mix

Illustration 12 : Substances utilisées pour le mix

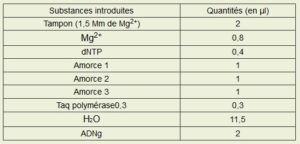
Tableau présentant la composition du mix* :
Explications de certains termes scientifiques :
- Mg2+ est un ion, celui du magnésium et permet, par sa présence, le bon déroulement de la réaction.
- Les dNTP sont les désoxynucléotides composant l’ADN (A,T,C et G) et lui permettant d’être allongé lorsqu’il sont présents dans le milieu.
- La taq Polymérase est une enzyme spécifique allongeant l’ADN à partir d’une amorce et de dNTP.
Définitions préalables pour comprendre la PCR :
- Amorce¹: Courte séquence d’ADN ou d’ARN complémentaire du début d’un brin matrice qui permet la synthèse du brin complémentaire de cette dernière matrice par une ADN polymérase.
- Polymerase²: Enzyme participant à la réplication de l’ADN. Une enzyme est par ailleurs une protéine permettant l’accélération des réactions chimiques de l’organisme.
- Désoxyribonucléotide⁴: groupement constitué d’une base nucléique (A,T,G ou C), d’un sucre le désoxyribose et d’un phosphate.
- Étape 1 (la dénaturation) :
Le fragment d’ADN est chauffé entre 94 et 96°C pendant une durée de 10 à 15 minutes. Les liaisons hydrogènes sont cassées et le double brin d’ADN est séparé en deux. Ainsi, nous passons d’un ADN double brin à un ADN simple brin. - Étape 2 (l’hybridation) :
L’appariement des amorces, cette étape dure entre 2 et 60 secondes et chauffe l’ADN à environ 68°C. Cela permet aux amorces¹ de s’hybrider (se fixer) à l’ADN simple brin. - Étape 3 (l’élongation) :
L’élongation dure entre 4 et 120 secondes (à 72°C) et permet aux polymérases²: de synthétiser le brin complémentaire à la matrice³. En effet, il y a ajout successif de désoxyribonucléotides⁴.
Illustration 13 : Schéma présentant les étapes de la PCR :
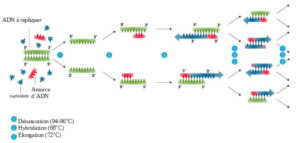
Notre PCR a été réalisée grâce à un thermocycleur* dans lequel nous avons placé nos tubes. Les étapes ont été répétées 30 fois chacune (30 cycles).
Le nombre de copies a été multiplié par 10⁹ du premier cycle au trentième. Durant l’expérience sont présents : la matrice ADN, les dNTPs, les polymérases et les amorces.
L’électrophorèse sur gel d’agarose
Le préfixe «électro» renvoie à l’électricité et la racine «phorèse» vient du grec «phoros» signifiant « porter d’un côté à l’autre ». Le terme «électrophorèse» décrit donc la migration de particules chargées sous l’influence d’un champ électrique. Il s’agit d’une méthode permettant, dans notre cas, d’étudier la composition de notre fragment d’ADN par comparaison afin de détecter la présence d’une modification génétique.
Tout d’abord, nous préparons le moule dans lequel nous allons déposer le mélange d’agarose (que nous allons réaliser). Pour cela, nous pesons 3 grammes d’agarose sur la balance ce qui nous donnera un gel de 3% (cf. illustration 7). Nous avons choisi un gel adapté au fragment d’ADN que nous voulions séparer. En effet, plus la solution est concentrée, plus elle séparera des fragments de petite taille. Nous avons mélangé le tampon avec l’agarose. Le tampon permet le maintien du pH quelles que soient les solutions que nous ajouterons. Elle a également ajouté un produit CMR afin que la molécule s’accroche à l’ADN le rendant visible (pour la photographie des résultats). Il s’agit d’un produit cancérogène, mutagène et reprotoxique étant donc dangereux pour la santé ce qui explique que nous ne l’avons pas manipulé (cf. illustration 9). Pour ceci, l’ingénieure portait des gants, une blouse et s’est placée sous une hotte. Une hotte est un dispositif permettant l’extraction des vapeurs toxiques provenant des produits utilisés lors des manipulations.
L’ingénieure a dissous le mélange en le passant au micro-ondes. Elle a ensuite agité l’erlenmeyer afin d’homogénéiser le mélange. Elle a également préparé le dispositif en plaçant les peignes dans la cuve électrophorétique (cf. illustration 8) puis y a déposé le gel. Nous avons ensuite laissé refroidir notre gel. Une fois refroidi, nous avons enlevé le peigne ce qui a permis de créer les puits dans lesquels nous déposerons nos fragments d’ADN. Ils se trouvent du côté négatif puisque l’ADN est chargé négativement. Puis, l’ingénieure a recouvert le gel d’un tampon d’électrophorèse.
Nous mélangeons désormais nos échantillons avec du bleu de bromophénol, solution bleutée permettant de les visualiser lorsque nous les déposerons dans le gel transparent. A l’aide d’une pipette de précision, nous entrons dans les puits verticalement et doucement afin de ne pas percer les puits (cf. illustration 10). Nous appuyons alors progressivement sur le piston de la pipette afin que les échantillons tombent au fond de ceux-ci. Nous refermons alors la cuve électrophorétique en mettant le couvercle puis nous branchons le générateur : le courant est lancé. Une tension va donc parcourir notre gel et le liquide inséré pour permettre la migration des fragments d’ADN dans le gel.
L’ADN est une molécule chargée négativement. Ainsi, dans un courant électrique, elle est attirée par la borne positive et se déplace selon son poids. Plus la bande d’ADN amplifiée est grande (donc plus elle est lourde) et moins elle migre vite. 30 minutes plus tard, nous arrêtons la migration en coupant le courant et récupérons notre gel.
Une fois que l’ADN a terminé de migrer, nous plaçons le gel sous un appareil photo ultraviolet pour voir le résultat. Une machine numérique a pris une photographie du résultat.
Photographies prises durant l’expérience :




Nos résultats et interprétations
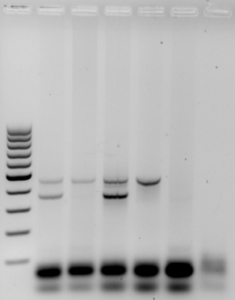
A la fin, grâce au gel d’électrophorèse, nous avons comparé les ADN à analyser à l’ADN «test» ce qui nous a permis de déduire la présence d’une modification du génome de l’organisme.
Tout à gauche, se situe la piste tour. Il s’agit d’un marqueur de poids moléculaire donnant une idée de la taille du fragment obtenu par PCR.
Ensuite, la première bande correspond à l’échantillon «test». Il s’agit d’un échantillon étant extrait d’un organisme dont nous savons qu’il a été muté génétiquement. Les autres bandes correspondent à nos quatre échantillons.
Lorsque nous observons une bande vers le haut, (échantillons 2 et 4) il s’agit d’échantillons – / – . Dans le troisième échantillon, une erreur de manipulation (due au dépôt de deux échantillons différents) a créé un mélange et faussé nos résultats.
Pour ce qui est du cinquième échantillon, nous n’observons pas de bande. Le liquide n’a donc pas été déposé dans le puits.
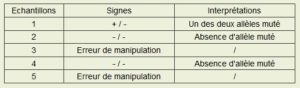
Tableau présentant les résultats de notre expérience :
Conclusion
Ainsi, nous pouvons affirmer que les allèles des échantillons 2 et 4 n’ont pas été mutés. Nous pouvons donc dire qu’ils n’ont pas été génétiquement modifiés contrairement à notre échantillon « test ». Pour ce qui est de nos échantillons 3 et 5, en raison d’une erreur de manipulation nous ne pouvons pas affirmer si les allèles ont été mutés ou non.